Rdzeniowy zanik mięśni (SMA) – objawy, przyczyny i leczenie rdzeniowego zaniku mięśni
Rdzeniowy zanik mięśni (SMA, z ang. spinal muscular atrophy) to choroba, która pojawiła się dość niedawno w katalogu badań przesiewowych skierowanych do noworodków. Mimo, że schorzenie jest bardzo poważne, wczesna diagnoza w zasadniczy sposób polepsza rokowania pacjenta. Czym tak naprawdę jest rdzeniowy zanik mięśni i czy należy się go obawiać?

Z różnych danych wynika, że jedno na 6000 – 9000 tysięcy dzieci rodzi się ze rdzeniowym zanikiem mięśni. Mimo, że statystycznie problem jest więc rzadki, stanowi on jedną z najczęściej wykrywanych wad genetycznych u małych dzieci. Niestety, należy też do najpowszechniejszych przyczyn śmierci wśród niemowląt.
Rdzeniowy zanik mięśni – co to za schorzenie?
SMA jest rzadkim schorzeniem genetycznym, które ma charakter wrodzony – nie można więc na nie po prostu zachorować, ani się nim zarazić. Sama nazwa wyjątkowo trafnie określa istotę choroby, które dotyka neuronów ruchowych zlokalizowanych w rdzeniu kręgowym, a odpowiedzialnych za pracę wielu kluczowych mięśni. Efektem tych zaburzeń jest atrofia, czyli stopniowe słabnięcie mięśni, które z kolei poważnie utrudnia codzienne funkcjonowanie.
Neurony ruchowe odpowiadają za poruszanie kończynami, klatką piersiową, językiem czy gardłem i mają zasadniczy wpływ na chodzenie, mówienie, oddychanie, czy przełykanie.
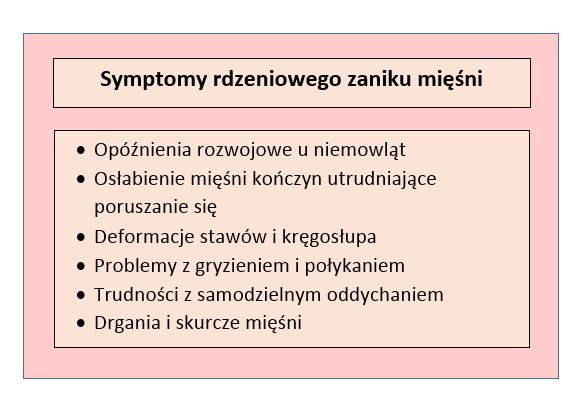
Objawy rdzeniowego zaniku mięśni
Istnieją cztery typu podstawowej formy rdzeniowego zaniku mięśni spowodowanego mutacją genu SMN1. Podstawą kategoryzacji jest wiek, w którym pojawiły się pierwsze symptomy – niestety, im wcześniej rozwija się choroba, tym dramatyczniejszy ma przebieg.
Typ 1 SMA, zwany również chorobą Werdniga – Hoffmana, to najgorzej rokująca postać schorzenia. Rozpoznaje się ją przed ukończeniem przez niemowlę 6 miesiąca życia, a typowe objawy obejmują zmniejszone napięcie mięśni, brak odruchów ze strony ścięgien, drgania mięśni, anomalie szkieletowe, a w najpoważniejszych przypadkach również chroniczne skrócenie mięśni i problemy z oddychaniem oraz przełykaniem. Bez leczenia mali pacjenci nie dożywają zwykle drugich urodzin.
Typ 2 diagnozowany jest u maluchów między 6 a 18 miesiącem życia. Potrafią one zwykle samodzielnie usiąść, ale nie są już w stanie nauczyć się stać czy chodzić. Mogą mieć problemy z oddychaniem. Większość pacjentów jednak przy odpowiednim leczeniu jest w stanie dożyć wieku dorosłego.
Typ 3 rozpoznawany jest po ukończeniu przez dziecko 18 miesięcy. Maluchy uczą się co prawda chodzić, ale miewają problemy z bieganiem czy wchodzeniem pod schody. Mimo problemów z kręgosłupem i skłonnością do chorób płuc ich oczekiwana długość życia nie jest jednak wyraźnie niższa od normalnej.
Typ 4 SMA diagnozowany jest dopiero po ukończeniu 21 lat i nie stanowi bezpośredniego zagrożenia dla życia. Większość pacjentów doświadcza „zaledwie” osłabienia mięśni nóg prowadzącego do częściowej utraty władzy w kończynach.
Tak naprawdę każdy przypadek rdzeniowego zaniku mięśni jest indywidualny, ale generalnie im później zacznie on dawać o sobie znać, tym lepsze rokowania. Warto podkreślić, że mimo poważnych często zaburzeń mięśniowych, chorzy cieszą się zwykle pełnym potencjałem emocjonalnym, mentalnym oraz zmysłowym.
Przyczyny rdzeniowego zaniku mięśni
Można powiedzieć, że rdzeniowy zanik mięśni jest problemem zakodowanym w DNA jednostki, najczęściej w chromosomie 5. Ściślej, choroba wynika z braku fragmentu kodu genetycznego, tzw. eksonu, w genie nazwanym SMN1. Dobra wiadomość jest taka, że sąsiadujący z nim gen SMN2 może częściowo zrekompensować straty wynikające defektu genetycznego – od liczby jego kopii zależy więc jak poważny przebieg będzie mieć SMA u danego człowieka.
W jaki sposób jeden pojedynczy gen może wpływać na cały organizm? SMN1 odpowiedzialny jest za produkcję białka SMN, bez którego neurony ruchowe nie mogą przetrwać. Istotą schorzenia jest więc tak naprawdę brak pojedynczej proteiny, która umożliwia przewodzenie impulsów nerwowych z rdzenia kręgowego do mięśni. Te ostatnie z powodu braku stymulacji słabną i kurczą się.
Aby problem zaistniał, dziecko musi odziedziczyć po jednej kopii wadliwego genu SMN1 od każdego z rodziców. W ogromnej większości przypadków są oni nieświadomi nosicielstwa defektu, bowiem posiadając tylko jeden zmutowany gen nie wykazują żadnych symptomów. W rzadkich wypadkach dziecko dziedziczy tylko jeden uszkodzony gen, a drugi podlega zjawisku mutacji podczas spontanicznego dzielenia się komórek w łonie matki.
Komplikacje rdzeniowego zaniku mięśni
Niestety, atrofia mięśni ma kolosalny wpływ na całokształt funkcjonowania organizmu. Pacjenci bardzo często mają problemy z poruszaniem się, siadaniem oraz chodzeniem i potrzebują na co dzień kul, chodzików czy nawet wózków inwalidzkich. Do częstszych komplikacji należą również:
- złamania kości, skrzywienia kręgosłupa i dyslokacje stawów;
- częste infekcje dróg oddechowych, w tym zapalenie płuc;
- niedożywienie wymagające sztucznego karmienia;
- zaburzenia funkcji oddechowej wymagające sztucznej wentylacji.
W niektórych przypadkach deformacja stawów jest tak silna, że pacjentom sugeruje się operacje, aby poprawić ich postawę lub poziom sprawności.
Badania przesiewowe w kierunku rdzeniowego zaniku mięśni
Diagnozę o SMA postawioną na bazie symptomów potwierdza się za pomocą testów z krwi (poziom kinazy kreatynowej) oraz elektromiografii (EMG), czyli badania określającego aktywność elektryczną nerwów oraz mięśni. Czasami dodatkowo przeprowadza się biopsję mięśni. Ponadto dziecku można wykonać test genetyczny z krwi, który pozwala zidentyfikować nieprawidłowości w zakresie genu SMN1. Coraz częściej oferuje się go noworodkom w ramach badań przesiewowych – wczesna identyfikacja choroby i rozpoczęcia leczenia znacząco poprawiają bowiem rokowania dziecka. Badania genetyczne pod kątem SMA przeprowadza się również na etapie ciąży, bądź to pod postacią odbioru wody płodowej (tzw. amniopunkcji), bądź też biopsji kosmówki. Pozwalają one określić defekt genetyczny już między 10 a 14 tygodniem ciąży.



Leczenie rdzeniowego zaniku mięśni
Jak dotąd nie znamy lekarstwa na SMA. W zależności od nasilenia symptomów chorym proponuje się szeroki wachlarz terapii wspomagających, które poprawiają komfort życia i przedłużają jego oczekiwaną długość. Bardzo dobre rezultaty przynosi na przykład rehabilitacja zamierzona na wzmacnianie poszczególnych funkcji, np. mówienia, połykania czy gryzienia. Zajęcia fizyczne mają na celu zapewnienie stawom elastyczności oraz opóźnienie procesu atrofii – tak, aby chory jak najdłużej cieszył się chociaż względną samodzielnością. Bardzo ważne są również nawyki żywieniowe, gdyż pacjenci z jednej strony muszą dbać o bogatą i zrównoważoną dietę, z drugiej, muszą jeść ostrożnie, aby uniknąć zadławienia spowodowanego niesprawnością mięśni odpowiadających za przełykanie.
Najważniejsze z punktu widzenia zatrzymania postępu choroby jest jednak podawanie leków nowej generacji, które odwracają część procesów wynikających z defektu genetycznego. W 2006 r. w Stanach Zjednoczonych zatwierdzono do użytku nusinersen (znany pod komercyjną nazwą Spinraza). Jego zadaniem jest zwiększanie produkcji białka SMN, kluczowe dla utrzymanie funkcjonalności neuronów ruchowych. Lek podaje się z dobrymi rezultatami dzieciom oraz dorosłym – w badaniach klinicznych u sporej części pacjentów zaobserwowano wyraźne zatrzymanie atrofii, a nawet jej częściowe odwrócenie.
Od 2019 roku stosuje się również terapię genową onasemnogene abeparovec-xioi (Zolgensma ™). Polega ona na wprowadzeniu do organizmu bezpiecznego dla człowieka wirusa, który przekazuje neuronom motorycznym w pełni funkcjonalny, zdrowy gen SMN. W rezultacie mięśnie mają lepszą stymulację i dłużej utrzymują się w formie. Terapia ta przeznaczona jest dla dzieci młodszych niż 2 lata.
Perspektywy dla chorych na rdzeniowy zanik mięśni
Dzięki rozwojowi medycyny pacjenci cierpiący na rdzeniowy zanik mięśni mają szansę na dłuższe i pełniejsze życie. Choć jesteśmy wciąż bardzo daleko od wyleczenia choroby, wczesna diagnoza i szerokie spektrum terapii pozwoliły osiągnąć niebywały postęp. Stąd tak wielu pediatrów zachęca do wykonywania przesiewowych testów genetycznych noworodków.
Nie ustają też próby opracowania nowych, skuteczniejszych leków. Kolejny obiecujący preparat na SMA wprowadzono do obrotu w USA w 2020 r., a dalsze są konsekwentnie rozwijane. Na dzień dzisiejszy nie ma jednak lepszej alternatywy niż uczestnictwo w wielokierunkowej terapii fizycznej i zajęciowej wspomagane zaleconymi przez lekarza preparatami.
- „Clinical Review Report: Nusinersen (Spinraza): . (Biogen Canada Inc.): Indication: Treatment of patients with 5q SMA” Canadian Agency for Drugs and Technologies in Medicine https://www.ncbi.nlm.nih.gov/books/NBK533981/, 3/03/2022;
- “Spinal Muscular Atrophy Information Page” NINDS, https://www.ninds.nih.gov/Disorders/All-Disorders/Spinal-Muscular-Atrophy-Information-Page, 3/03/2022;
- “Spinal Muscular Atrophy (SMA)” MDA, https://www.mda.org/disease/spinal-muscular-atrophy/research, 3/03/2022;
- “Spinal Muscular Atrophy (SMA)” Johns Hopkins Medicine, https://www.hopkinsmedicine.org/health/conditions-and-diseases/spinal-muscular-atrophy-sma, 3/03/2022;
- “Spinal muscular atrophy” NHS, https://www.nhs.uk/conditions/spinal-muscular-atrophy-sma/, 3/03/2022;
- “Spinal Muscular Atrophy” Mary Jo DiLonardo, https://www.webmd.com/brain/spinal-muscular-atrophy#1, 3/03/2022;


















